《分子科学学报》

当一个分子被光击中时,会发生什么?这或许是
来源:分子科学学报 【在线投稿】 栏目:综合新闻 时间:2021-10-13金属配合物在与光相互作用中表现出令人着迷的行为,例如用于有机发光二极管、太阳能电池、量子计算机,甚至用于癌症治疗。在许多这样的应用中,电子自旋,一种电子的固有旋转,起着重要的作用。现在维也纳大学化学学院的化学家塞巴斯蒂安·马伊和莱蒂西亚·冈萨雷斯,成功地模拟了由金属配合物的光吸收引发的极快自旋翻转过程,其研究发现发表在《化学科学》期刊上。当一个分子被光击中时,在许多情况下会引发所谓的“光诱导”反应。
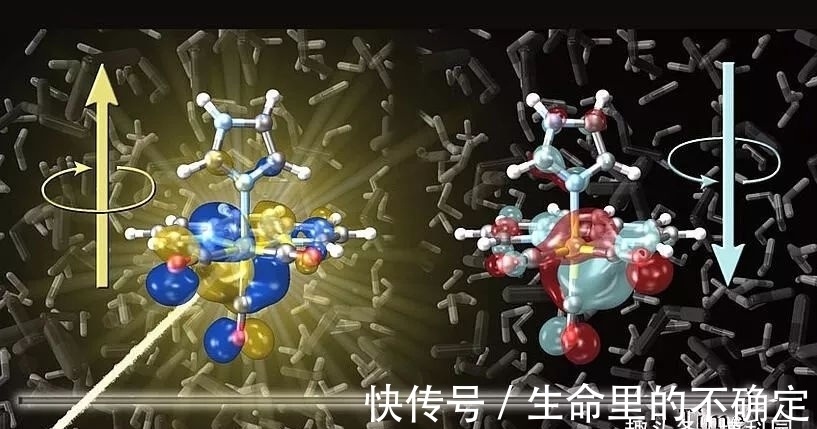
这可以被认为是电子运动和核运动的相互作用。首先,光的吸收充满能量地“激发”电子,例如可以削弱一些键。随后,重得多的原子核开始移动。电子可以从一个轨道切换到另一个轨道。在“自旋-轨道耦合”的物理效应的控制下,电子自旋可以在同一时刻翻转。这种运动的相互作用就是为什么分子中的自旋翻转过程,通常需要相当长的时间。然而,计算机模拟表明,在某些金属配合物中并非如此,例如,在被检查的铼复合物中,自旋翻转过程已经在10飞秒内发生。
即使在这段短时间内原子核实际上是静止的,即使光在这段时间内移动也只有千分之三毫米。这对于精确控制电子自旋特别有用,例如在量子计算机中。研究过程中最大的困难之一是模拟需大量计算机能力,虽然对于有机小分子来说:现在已经可以用适当的计算量进行非常精确的模拟,但金属配合物提出了更大的挑战。除其他原因外,这是由于大量的原子、电子和溶剂分子需要包括在模拟中,但也因为电子自旋只能用相对论的方程来精确描述。
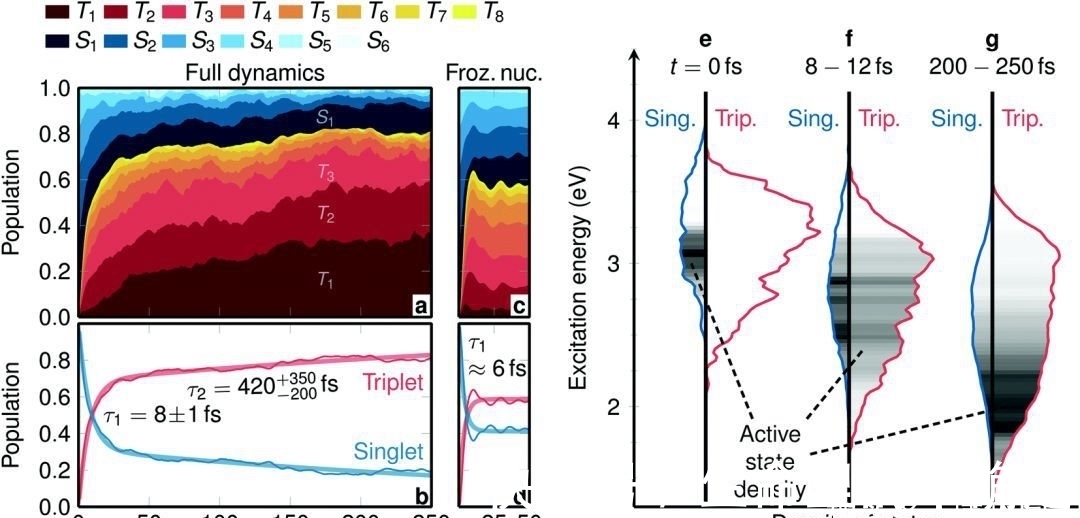
理论化学研究所的科学家们在研究过程中总共在奥地利超级计算机“维也纳科学集群”上花费了近一百万个计算机小时,这相当于一台典型个人计算机上大约100年的计算机时间。分子自旋的变化在化学和生物学中普遍存在,在自旋翻转过程中,最快的过程之一是过渡金属配合物中的系统间交叉(ISC)。在研究中,使用了显式水溶液中广泛的全维激发态动力学模拟研究了[Re(CO)3(Im)(Phen)]+(im=咪唑,phen=邻菲咯啉)的自旋弛豫动力学和发射光谱。

与在其他过渡金属配合物中观察到的相反,从单重态到三重态的转变通过两步过程发生,具有两个不同时间尺度的明显可分离电子和核驱动组件。这个ISC过程比以前从发射光谱中记录的这个和其他铼(I)羰基二亚胺配合物快一个数量级。包括显式激光场相互作用在内的模拟证明,需要几个周期的紫外激光脉冲来跟踪这种分子自旋轨道波包的产生和演化。动力学分析还揭示了延迟ISC组分,这可以用分子内振动能量重新分布来解释。这些结果为过渡金属配合物的超快体系间交叉提供了基础认识。
博科园|研究/来自:维也纳大学
参考期刊《化学科学》
DOI: 10.1039/C9SC03671G
博科园|科学、科技、科研、科普
关注【博科园】看更多大美宇宙科学哦